
The purpose of the eukaryotic cell cycle is to accurately duplicate and segregate the genome. Progression through the cell cycle is dependent on a class of proteins called the cyclins and their associated cyclin dependent kinases (cdks). The activity of these reversible switches regulates cell cycle progression to ensure that a DNA synthesis phase (S-phase) always alternates with a chromosome segregation phase (M-phase). With a few notable exceptions, this rule is followed by all dividing eukaryotic cells and a failure to follow this rule results in either cell death or the generation of disease states such as cancer. In addition, all eukaryotic cells have checkpoint pathways that inhibit cell cycle progression in the presence of incompletely replicated DNA or DNA damage. These checkpoint kinases inhibit cell cycle progression by inhibiting the activity of the cyclin/cdk complex. One such complex, cyclin B/cdk1 is required for entry into mitosis in mammalian cells.
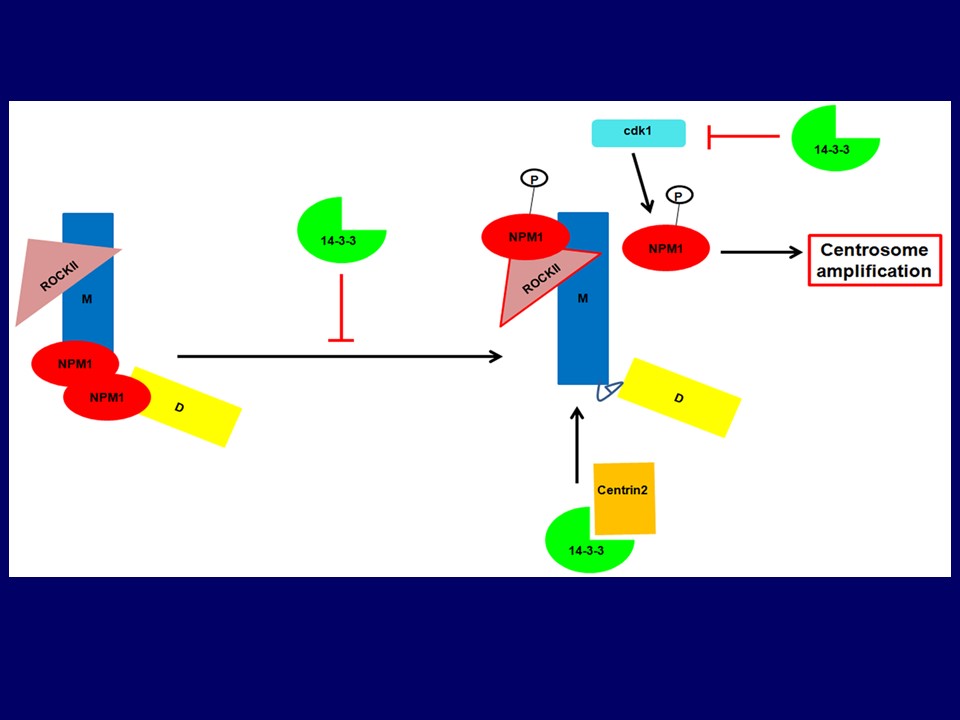
The dual specificity phosphatase, cdc25C, activates the cyclin B/cdk1 complex leading to mitotic progression. The activity of cdc25C is inhibited during interphase and upon checkpoint activation through complex formation with the 14-3-3 protein family. The 14-3-3 proteins are a family of conserved proteins that bind to proteins containing a phosphorylated Serine or Threonine residue in one of two consensus motifs. Previous results from our laboratory have demonstrated that loss of 14-3-3ε and 14-3-3γ result in the activation of cdc25C and premature cell cycle progression in the presence of DNA damage and incomplete S-phase. We have recently demonstrated that loss of 14-3-3γ leads to an increase in centrosome number in cells. This can be reversed by over expression of an shRNA resistant 14-3-3γ cDNA. The increase in centrosome number leads to genomic instability and aneuploidy with passage in culture, which is accompanied by increased colony formation in soft agar and tumor formation in the mouse. To identify the molecular mechanism underlying this increase in centrosome number, we demonstrated that over expression of cdc25 family members exacerbated the centrosome phenotype while a knockdown of cdc25 family members resulted in an inhibition of centrosome duplication. These results could also be phenocopied by over expression of the constitutively active cdk1AF mutant that cannot be inhibited by phosphorylation. This also required an increase in cdk1 activity and was accompanied by an increase in the phosphorylation of the centrosomal cdk1 target, NPM1. We have further demonstrated that the increase in centrosome number is accompanied by an increase in centrosome clustering over passage thus resulting in an increase in aneuploidy. Complete disruption of the 14-3-3-cdc25C complex led to an increase in the number of cells with multi-polar spindles and a decrease in centrosome clustering. This was accompanied by a decrease in colony formation in soft agar and a decrease in tumor formation in nude mice. Our results suggest that premature activation of cdk1 in interphase cells leads could lead to tumor cell killing in vivo and that one way to activate cdk1 function is by disrupting the cdc25C-14-3-3 complex. Our results have demonstrated that disrupting the cdc25C-14-3-3 complex could lead to tumor cell killing along with a decrease in centrosome clustering. We are developing imaging assays to identify drugs that will disrupt clustering in these cells with the hope that these will serve as lead compounds for the treatment of neoplastic disease. In addition, we would like to continue our research on how 14-3-3 proteins regulate centrosome formation as these results will tell us more about the biology of this important organelle in addition to identifying potential 14-3-3 targets that might be involved in centrosome assembly and maturation.