IPD 2.0: To derive insights from an evolving SARS-CoV-2 genome
Desai S, Rane A, Joshi A, Dutt A.Here, we present an updated version of our automated computational pipeline, Infectious Pathogen Detector (IPD) 2.0 with a SARS-CoV-2 module, to perform a comparative genomic analysis to better understand the pathogenesis and virulence of the virus. This manuscript extends, complements and updates our previous study (Brief Bioinform, 2021 Jan 22:bbaa437.doi: 10.1093/bib/bbaa437), wherein we described 4634 SARS-CoV-2 mutations identified from few thousand genomes with distinct hotspot mutations in the S gene.
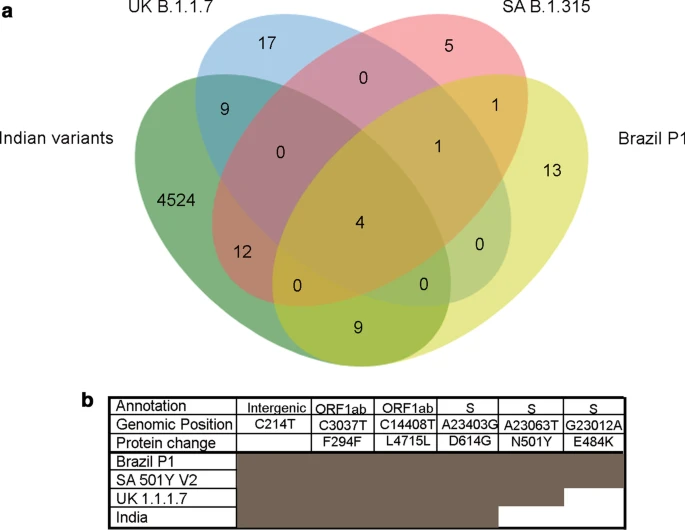
We describe the additional features and improvements in the IPD 2.0 over its predecessor. With the increasing submissions of SARS-CoV-2 genomes to a public database, it becomes imperative to integrate the updated strain and variant information during analysis. To address this issue, we have developed an automated module for upgrading the SARS-CoV-2 variant database of IPD 2.0, making the tool contemporary and dynamic. Through the automated analysis of the update module of the IPD 2.0 SARS-CoV-2 module, we present the analysis of 2.58 million SARS-CoV-2 mutations found in 208,911 samples from 155 different countries from GISAID. From the variants obtained analyzing 208,911 samples, we find 1,004,453 (38.88%) synonymous, 1,327,548 (51.39 %) nonsynonymous mutations and 242631 (9.39%) mutations in the intergenic region with an overall 6.6 nonsynonymous and 5 synonymous mutations per sample. Our analysis reveals that after normalizing for gene length, the S, N, M, ORF7a, and ORF10 viral genes were selectively enriched in nonsynonymous mutations compared to synonymous mutations that comprised 54.36% of all SARS-CoV-2 nonsynonymous mutations. We further analyzed the emerging predominant strains, B 1.1.7 (UK), B 1.135 (South Africa), P1 (Brazil), which revealed the private as well as shared mutations among these strains. Comparing the recurrent mutations in these three strains with the variants in the Indian samples, we found various distinctly shared mutations across Indian and other emerging strains. However, no specific incidence of N501Y and E484K mutations in the S protein was observed in the Indian samples.
Thus, we update IPD2.0 with an extensive database of sample-wise variants and clade annotation, which forms the core of the SARS-CoV-2 analysis module of the analysis pipeline. In addition to the improvements in speed and portability of the IPD 2.0 pipeline, we demonstrate the clade assignment accuracy of the SARS-CoV-2 module to be 92.8 % using a simulated dataset. We further explore the effect of coverage, the number of variants per sample and background mutation rate on the clade prediction accuracy of IPD 2.0.