
Integrated genomic characterization of head and neck cancer cell lines of Indian origin
Several large scale cancer genome sequencing projects involving large sample sets have identified spectrum of driver genomic alterations. Statistical methodologies like MutSig, Genome MuSiC, Intogen, InVEx, ActiveDrive and GISTIC aid in identifying significantly altered genes across large sample cohorts by comparing rate of mutations of each gene with background mutation rate to determine an unbiased enrichment. However, these elegant tools are not applicable for studies involving fewer or rare clinical specimen that are otherwise inherently restrictive due to the limited statistical power to detect alterations existing at lower frequency. An alternative to data-type studies with fewer clinical specimen is to combine multiple data types assessing variation across multiple levels of biological regulation in the same sample, with an estimation of overall higher cumulative effect outcome. Combinatorial sources of genetic evidence converging at same gene can limit false positives by filtering strategy and potentially reducing the multiple hypothesis testing burden for identification of causal genotype-phenotype association. We present here for the first report detailing cytogenetic characterization, microarray analysis, whole exome and transcriptome sequencing of four HNSCC-derived cells lines established from Indian head and neck cancer patients. We applied the widely used posterior filtering strategy of results obtained from genome wide studies to effectively reduce the amount of data obtained from individual platforms. Adopting such an integrative approach allow us to identify biological relevant alterations affected by two or more events even from fewer samples. Besides identifying major HNSCC hallmark genes, we find a driver heterozygous truncating mutation in Nuclear receptor binding protein NRBP1 (Q73*), identical to as reported in other cancers and similar to activating mutant alleles in non-human model organisms. We provide the first functional analysis of mutant NRBP1 and establish that NIH3T3 cells expressing the mutant NRBP1 enhance their survival and anchorage independent growth, consistent with its suggestive role in prostate cancer biology and other model organisms.
Next, we asked if computation of cumulative frequency of genes known to be altered in TCGA HNSCC dataset may provide insights to identify additional genes with crucial role based on the integrated analysis. We identify three most significantly altered candidate novel oncogenes-- UBR5, ZNF384 and TERT altered at 32%, 19%, and 16%, respectively that has not been previously described in HNSCC.
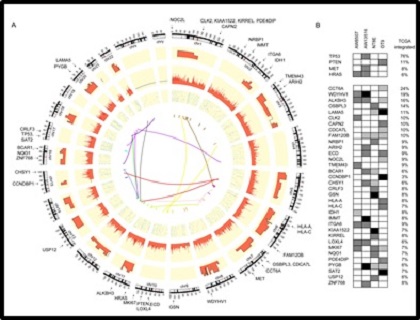
[Integrative genomic landscape of HNSCC. (A) Circos plot representations of genes identified by integrated genomic analysis of HNSCC cell lines. From outside to inside: karyotype, CNVs, Gene expression (FPKM), SNVs and translocations. (B) Heatmap representation of novel genes identified by integrated analysis of four HNSCC cell lines and their incidence in 279 HNSCC samples from TCGA study.]
[BMC Genomics]