Running TMC-SNPdb 2.0 toolkit
The required scripts to run TMCSNPdb 2.0 toolkit are placed in the “src” directory.
To load the R Shiny based GUI or via web-browser use the following command:
In Linux terminal:
xxxxxxxxxx$ conda activate tmcsnpdb2tk$ R -e "shiny::runApp('~/tmcsnpdb2tk/app.R')"In Windows CMD:
xxxxxxxxxx> "Path/to/R.exe" -e "shiny::runApp("~/tmcsnpdb2tk/app.R", launch.browser = TRUE)"
Guide to use TMC-SNPdb 2.0 toolkit
Once users executes the above commands TMCSNPdb 2.0 toolkit GUI appears with 3 tabs.
Following are the detailed description for each of the tabs-
dbCreator
Allows user to create custom germline database from individual .vcf or .gz (bgzip compressed) files.
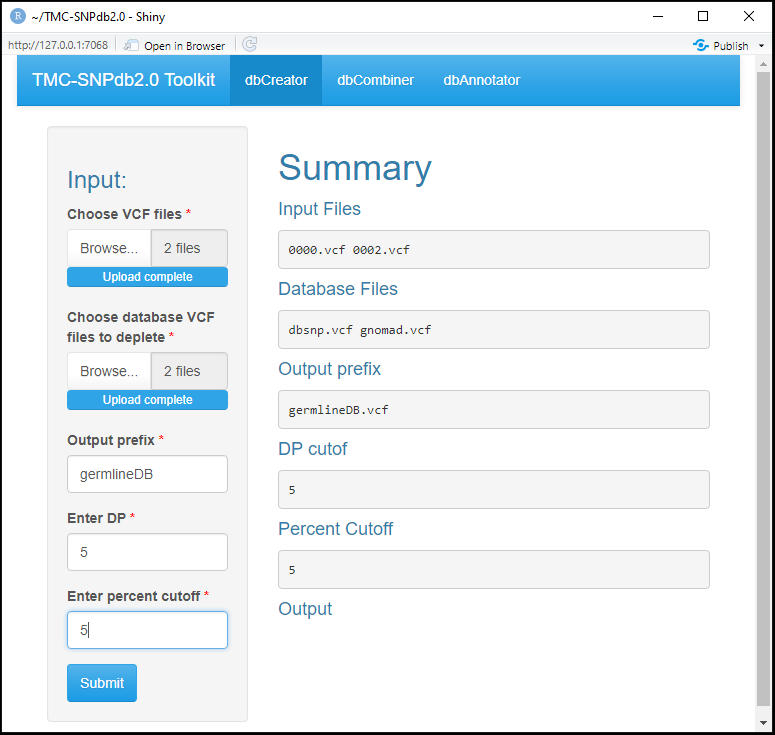
Files generated in steps 3 and 4 in pre-processing steps can be uploaded here which will combine all the variants and will generate merged.vcf file and further will apply filter of DP that is maximum 5 reads supporting the variant position (DP=5) and percent cutoff to check the recurrence of the variants in 5 percent of samples. Further these variants will deplete against various public databases which is provided in "Choose database VCF files to deplete" (database such as dbSNP,gnomAD,GenomeAsia 100K,Indigenomes,TMC-SNPdb 2.0) which will result the unique variants which will be specific to population which is not present in any of the mentioned databases.
dbCombiner
Allows user to combine multiple .vcf.gz (bgzip compressed) database files with TMC-SNPdb 2.0 database.
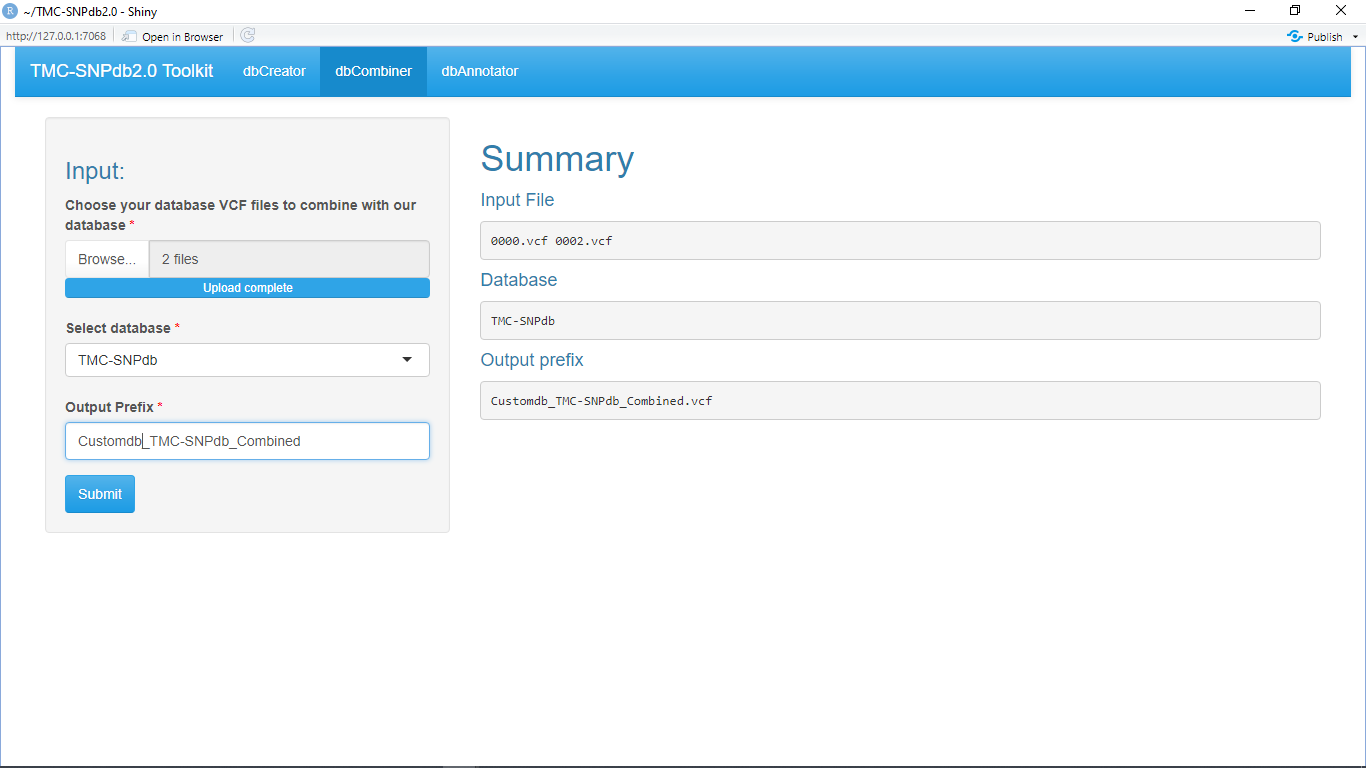
Database generated by dbCreator can be used as a input in dbCombiner (other bgzip compressed vcf database) can also be provided as a input to merge with our individual as well as combined ethnic database (TMC-SNPdb 2.0 [1], IndiGenomes [2], GenomeAsia 100K [3])
dbAnnotator
Allows user to annotate and flag multiple .vcf files for the presence of variants in TMC-SNPdb 2.0 database.
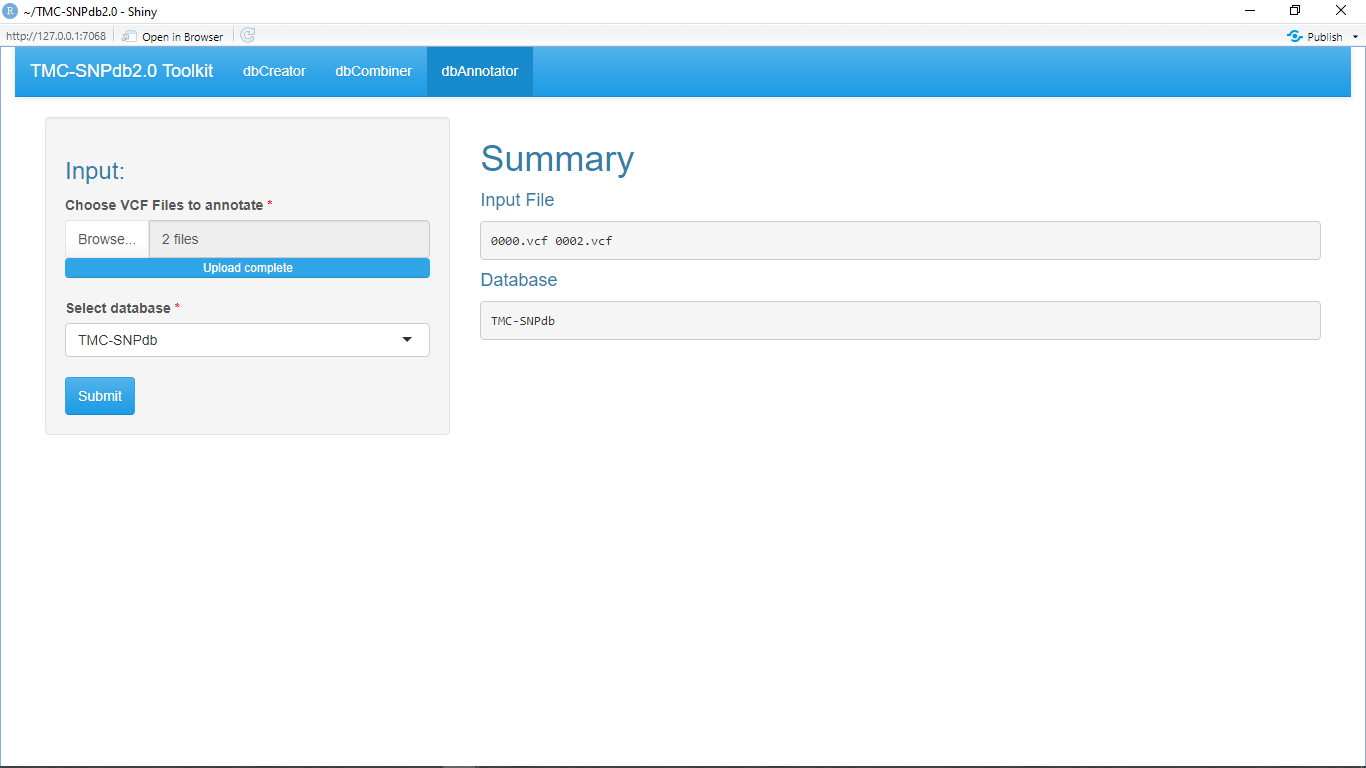
dbAnnotator flags in INFO column if the variant is present in any of our databases (TMC-SNPdb 2.0 [1], IndiGenomes [2], GenomeAsia 100K [3]) for any given vcf file.
TMC-SNPdb 2.0 toolkit output
dbCreator generates two output files:
- Outputs merged.vcf file which contains all the variants from individual .vcf files
- Outputs output_prefix.vcf which is obtained after applying filter of DP cut-off as well as presence of variants in 5 percent of samples from merged.vcf
dbCombiner creates following output files:
- Outputs combined.vcf file which contains the variants from TMC-SNPdb 2.0 database as well as from the custom database.
dbAnnotator creates following output files:
- Outputs _annotated.vcf file which flags the presence of variants in TMC-SNPdb 2.0 database.